Un gruppo di ricercatori della Scuola internazionale Superiore di Studi avanzati (SISSA) di Trieste e dell’Università di Cambridge ha escogitato un metodo che riduce i tempi per simulare come le proteine assumono le caratteristiche forme tridimensionali. Si tratta di un’informazione importante per comprendere la loro funzione, che oggi viene normalmente ricavata con tecniche sperimentali spesso molto costose.
Un gruppo di ricercatori della Scuola internazionale Superiore di Studi avanzati (SISSA) di Trieste e dell’Università di Cambridge ha escogitato un metodo che riduce i tempi per simulare come le proteine assumono le caratteristiche forme tridimensionali. Si tratta di un’informazione importante per comprendere la loro funzione, che oggi viene normalmente ricavata con tecniche sperimentali spesso molto costose.
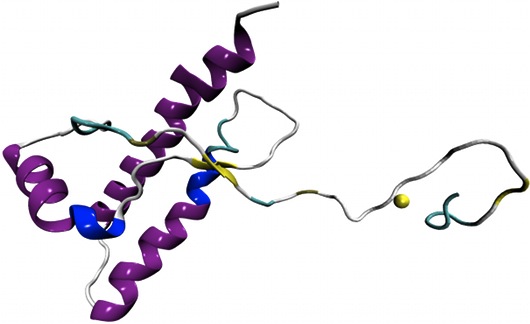
Per capire come funzionano le proteine è importante conoscere la loro forma tridimensionale, ma anche come questa si viene a creare. Bisogna cioè capire in che modo il filamento di aminoacidi che compone le proteine si ripiega su se stesso per assumere una forma specifica.
Lo studio della dinamica molecolare delle proteine si basa oggi su tecniche di simulazione al computer in cui il sistema viene trattato come un insieme tridimensionale di palline (1 pallina = 1 atomo), che viene osservato mentre evolve nel tempo. Questa tecnica è molto accurata, ma piuttosto lenta, e un gruppo di ricercatori fra cui Daniele Granata e Alessandro Laio della SISSA di Trieste ha escogitato un trucco per ridurre i tempi di simulazione.
“Sfruttiamo i dati sperimentali ottenuti osservando le proteine con la risonanza magnetica nucleare, e con questi creiamo dei vincoli da applicare al modello”, ha spiegato Laio, che ha coordinato la ricerca pubblicata sui Proceedings of the National Academy of Sciences (PNAS).
“In pratica abbiamo usato un ‘trucco’. Provate a immaginare che io vi tiri per un braccio verso un certo posto, diciamo, per aver un riferimento, da Trieste verso Rimini. Con il trucco si impiega un tempo anche 1000 volte inferiore al tempo normale richiesto per quel tragitto. Grazie a regole matematiche – dedotte dalle osservazioni precedenti sui ‘viaggi a Rimini’ – posso calcolare, partendo dal tempo di percorrenza truccato, quanto lo stesso individuo ci avrebbe messo per arrivare spontaneamente nello stesso posto, senza essere tirato. Lo stesso tipo di ragionamento si può fare su una proteina che deve ripiegarsi per raggiungere una certa forma”.
“C’è una legge che lega il tempo che la proteina ci mette per ripiegarsi col ‘trucco’ e quello che ci mette per farlo spontaneamente”, spiega Laio.
La tecnica adottata da Granata e Laio, applicata in questo caso alla proteina G del batterio streptococco, ha permesso di ottenere risultati perfettamente coerenti con quelli della tecnica più comune, ma con il vantaggio di accorciare notevolmente i tempi di calcolo. “Data la lentezza delle nomali tecniche di dinamica molecolare al computer, metodi come il nostro ottimizzano i tempi di elaborazione, e potrebbero dare un grosso impulso alla ricerca”.